Publications
Publications
 Partners
Partners



BACKGROUND
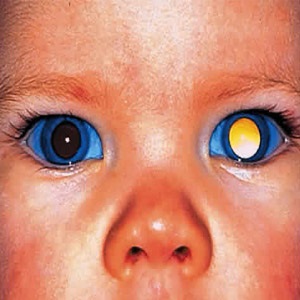
Small tumors develop in the retina, which is located at the back of the eye. The retina sends visual images to the brain where they are perceived. These tumors may cause the pupils to appear white instead of black. Although most people only have retinoblastoma in one eye, sometimes tumors develop in both eyes.
About 25% of cases are inherited or passed down from parents to their children. The other 75% of cases are caused by a random genetic mutation that occurs during the development of the egg, sperm, or embryo.
If left untreated, retinoblastoma may be fatal. This is because tumors may spread to the brain via the optic nerve. Brain tumors, called pinealoma, are most likely to occur in the hereditary form of retinoblastoma. In addition, people with hereditary retinoblastoma have an increased risk of developing bone cancer (called osteosarcoma), soft tissue cancers, melanoma, and other types of cancer.
Factors, such as age, gender, race, and ethnicity, do not increase a person's risk of developing retinoblastoma. On average, retinoblastoma affects about one person out of 18,000-30,000 live births worldwide. Researchers estimate that about one out of 15,000-16,000 people are born with retinoblastoma in both the United States and northern Europe. An estimated 250-500 new cases of retinoblastoma are diagnosed each year in the United States.
Retinoblastoma may be treated with radiation, chemotherapy, and/or surgery. Sometimes the eye may need to be removed. The cancer is successfully treated in 86-93% of cases. If the condition is effectively treated, people can expect to live long, healthy lives.
CAUSES
Mutations in the RB1 gene: People who are born with a mutated RB1 gene have an increased risk of developing retinoblastoma. These mutations are responsible for most cases of retinoblastoma. Normally, this gene helps prevent cells from growing and dividing rapidly or uncontrollably. Most mutations in the RB1 gene result in uncontrolled cellular growth, which leads to the development of tumors in the eyes.
Mutations in the RB1 gene are inherited as an autosomal dominant trait, which means that one copy of a mutated gene in each cell is enough to increase a person's risk of developing retinoblastoma. Each gene has two variations, called alleles. One allele is inherited from each parent. A person with retinoblastoma may inherit one copy of the gene or it may develop randomly during the formation of the egg, sperm, or embryo. In order for retinoblastoma to develop in the person, a second mutation in the other copy of the RB1 gene must occur randomly in the retinal cell during a person's lifetime.
If a person has a family history of retinoblastoma or if tumors develop in both eyes, the mutated RB1 gene is probably in all of the person's cells, including sperm or egg cells. Such people have what is called hereditary retinoblastoma. These individuals may pass the mutated gene onto their children.
If there is no family history of retinoblastoma and only one eye is affected, the mutated RB1 gene may only be in the tumor cells. In such cases, the disease is most likely the non-inherited form of retinoblastoma. Family members of people with non-inherited retinoblastoma do not have an increased risk of developing the condition.
Partial deletion of chromosome 13: A minority of retinoblastoma cases occur when the part of chromosome 13 that contains the RB1 gene is deleted. This specific part of the chromosome is commonly called 13q14. When this part of the chromosome is deleted, other symptoms, such as mental retardation and delayed growth, may occur.
Most deletions of 13q14 are not inherited. Instead, the deletion occurs randomly during the formation of the egg, sperm, or embryo.
SIGNS AND SYMPTOMS
The pupil of the eye may be white, instead of black. This may be apparent to the naked eye. It is especially noticeable in photographs that are taken with a flash camera. Some people may also have lazy eyes (called strabismus), which occurs when the eyes do not appear to be looking in the same direction. Less common symptoms may include reddening of the white part of the eye, pain, vision problems, and a pupil that does not contract when it is exposed to light.
People with partial deletions of chromosome 13 may experience additional symptoms, such as mental retardation, distinct facial features (such as prominent eyebrows, a small nose with a broad nasal bridge, and ear abnormalities), and delayed growth.
DIAGNOSIS
General: If retinoblastoma is suspected, an eye doctor, called an ophthalmologist, will examine the child's eyes. In most cases, the condition can be diagnosed after looking at the eye with special magnifying lenses and lights. Sometimes an imaging study, such as an ultrasound, computerized tomography (CT) scan, or magnetic resonance imaging (MRI) scan, may be performed to determine how large the tumor is and if it has spread to other parts of the body.
Ultrasound: If the tumor is so big that the ophthalmologist is unable to see the entire inside of the eye, an ultrasound may be used.
Computerized tomography (CT): A computerized tomography (CT) scan takes pictures of the inside of the eye. Before a CT, a dye might be injected into the patient's blood. The pictures provide detailed images of the eyes, including the size and location of any tumors that might be present.
Magnetic resonance imaging (MRI): A magnetic resonance imaging (MRI) scan provides more detailed images than a CT scan, but it takes longer to perform. During an MRI, the patient is required to lie completely still while the machine takes pictures of the eyes. For this reason, some young children may receive sedatives before an MRI.
Genetic testing: People with family histories of retinoblastoma may choose to undergo genetic testing. If a person has a mutated gene associated with retinoblastoma he/she has an increased risk of developing the disorder in the future. Therefore, he/she is encouraged to undergo regular screenings for the disorder. Genetic testing may also help determine if a person has the type of retinoblastoma that can be passed down to his/her children.
COMPLICATIONS
General: People that have hereditary retinoblastoma have an increased risk of developing various types of cancers. Therefore, people with retinoblastoma are encouraged to visit their doctors regularly to monitor their conditions. They should also undergo regular cancer screenings, such as mammograms and skin cancer tests. Below are some of the most common cancers associated with this condition.
Osteosarcoma: People with hereditary retinoblastoma may develop osteosarcoma, which is a type of bone cancer that usually affects the arms and legs. It typically causes pain and swelling. Prompt treatment is necessary because it may spread to other parts of the body.
Malignant melanoma: People with hereditary retinoblastoma may develop an aggressive type of skin cancer called malignant melanoma. If diagnosed early, malignant melanoma is treatable. However, if it is left untreated, the cancer may spread to other parts of the body. Once the cancer has spread, it is difficult to treat and it may be fatal.
Soft tissue sarcomas: People with hereditary retinoblastoma have an increased risk of developing soft tissue sarcomas. These are cancers that develop in the muscles, tendons, ligaments, and fatty tissues of the body. Like other types of cancer, soft tissue sarcomas may spread to other parts of the body, including the organs, if left untreated.
Less common types of cancer: Less common types of cancer associated with hereditary retinoblastoma include brain tumors, lymphoma, breast cancer, and lung cancer.
TREATMENT
General: On average, retinoblastoma is successfully treated in 86-93% of cases. Treatment for retinoblastoma depends on many factors, including the size and location of the tumor, as well as whether one or both eyes are affected. Treatment options include radiation, chemotherapy, and/or surgery. If only one eye is affected, the tumor may cause permanent vision damage. In such cases, the eye is surgically removed. Patients receive treatment from specialized doctors called ophthalmic oncologists.
External radiation: External-beam radiation therapy may be used to kill cancer cells in the retina. A machine is used to focus radiation on the retina. This procedure is typically performed at a hospital about five times a week for three or four weeks. Each session lasts a few minutes.
Common side effects include fatigue, skin irritation near the eyes, decreased appetite, and inflammation of the tissues near the eye. Radiation therapy may also inhibit the growth of bone and other tissues near the eye. It may increase the patient's risk of developing other types of cancers.
Internal radiation: Internal radiation may be used to treat small tumors caused by retinoblastoma. An operation is performed to temporarily insert radioactive material inside the patient's eye socket. The radioactive material is usually kept inside the eye for four to five days. Then the implant is surgically removed.
Side effects of internal radiation are the same as external radiation.
Laser therapy: Laser therapy may also be used to kill small tumors in the eyes and/or to kill blood vessels that feed the tumors. This therapy is typically repeated once a month for as long as three months.
Freezing (cryotherapy): A type of therapy called cryotherapy may be used to treat small tumors caused by retinoblastoma. The procedure is performed at the hospital, and the patient receives general anesthesia. An extremely cold probe is placed next to the tumor to kill the cancer cells. The eye and eyelid may be swollen for a few days after the procedure. Cryotherapy is typically repeated several times before the tumor is successfully killed.
Chemotherapy: Some patients may receive chemotherapy drugs that help kill or slow the growth of cancer cells. Some drugs are taken by mouth, while others are injected into a vein using a syringe or an intravenous (IV) drip device. Because these drugs affect the entire body, they may help treat cancers that have spread beyond the eyes. However, retinoblastoma is often resistant to chemotherapy. Therefore, different combinations of drugs may need to be used to effectively treat the cancer.
Common side effects of chemotherapy include nausea, vomiting, fatigue, hair loss, anemia, confusion, depression, problems with blood clotting, sores in the mouth (called stomatitis), sores in the throat (called mucosititis), dry mouth, diarrhea, constipation, loss of appetite, peripheral neuropathy (which causes burning, weakness, tingling, or numbness in the hands and/or feet), acne, dry skin, rash, yellow and brittle nails, flu-like symptoms, fluid retention, decreased sperm motility, and reduced sexual hormone production in women. Some chemotherapy drugs may also damage the kidneys and/or bladder. Chemotherapy destroys healthy immune cells. Therefore, patients undergoing chemotherapy have weakened immune systems and are susceptible to infections.
Surgical removal of the eye (enucleation): If only one eye is affected, the tumor may cause permanent vision damage. In such cases, the eye is surgically removed. After surgery, an artificial eye (called an ocular prosthesis) is implanted as a cosmetic substitute for the real eye.
INTEGRATIVE THERAPIES
Currently, there is a lack of scientific data on the use of integrative therapies for the treatment or prevention of retinoblastoma.
PREVENTION
There is currently no known method of prevention against retinoblastoma. However, people with family histories of retinoblastoma may choose to undergo genetic testing. If a person has a mutated gene associated with retinoblastoma he/she has an increased risk of developing the disorder later in life. Therefore, he/she is encouraged to undergo regular screenings for the disorder. Genetic testing may also help determine if a person has the type of retinoblastoma that can be passed down to his/her children.
Early diagnosis and prompt treatment may help reduce the risk of permanent vision loss.
AUTHOR INFORMATION
This information has been edited and peer-reviewed by contributors to the Natural Standard Research Collaboration (www.naturalstandard.com).
- Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25;1:31. View abstract
- Chintagumpala M, Chevez-Barrios P, Paysse EA, et al. Retinoblastoma: review of current management. Oncologist. 2007 Oct;12(10):1237-46. View abstract
- De Potter P. Current treatment of retinoblastoma. Curr Opin Ophthalmol. 2002 Oct;13(5):331-6. View abstract
- Doz F. Retinoblatoma: a review. Arch Pediatr. 2006 Oct;13(10):1329-37. Epub 2006 Aug 23. View abstract
- Finger PT, Czechonska G, Demirci H, et al. Chemotherapy for retinoblastoma: a current topic. Drugs. 1999 Dec;58(6):983-96. View abstract
- Kinge B, Tranheim RS, Eide NA. Retinoblastoma—hereditary eye cancer in children. Tidsskr Nor Laegeforen. 2004 Jan 22;124(2):183-5. View abstract
- National Cancer Institute (NCI). www.cancer.gov. Accessed December 6, 2007.
- National Eye Institute (NEI). www.nei.nih.gov. Accessed December 6, 2007.
- National Human Genome Research Institute (NHGRI). www.genome.gov. Accessed December 6, 2007.
- Natural Standard: The Authority on Integrative Medicine. www.naturalstandard.com. Copyright © 2008. Accessed December 6, 2007.
Copyright © 2011 Natural Standard (www.naturalstandard.com)